本文刊于:中华儿科杂志, 2023,61(12) : 1077-1085
DOI: 10.3760/cma.j.cn112140-20230904-00153
作者:中华医学会儿科学分会神经学组 福棠儿童医学发展研究中心神经内科专业委员会 中华儿科杂志编辑委员会
通信作者:方方,Email:fangfang@bch.com.cn;姜玉武,Email:jiangyuwu@bjmu.edu.cn
摘要
Leigh综合征是儿童期最常见的线粒体脑肌病。多于婴幼儿期起病,临床常有发育迟缓、发育倒退等症状;影像学检查可见双侧对称的脑干和(或)基底节病变。Leigh 综合征的确诊主要依赖基因检测,组织病理和酶学检测可进一步协助诊断。目前,国内Leigh综合征的诊断和治疗尚缺乏规范,因此中华医学会儿科学分会神经学组、福棠儿童医学发展研究中心神经内科专业委员会和中华儿科杂志编辑委员会联合发起制订本共识。本共识使用德尔菲方法制订,对Leigh综合征的诊断和治疗提出24条推荐意见。
Leigh综合征(OMIM #256000)又称亚急性坏死性脑脊髓病,是儿童期最常见的线粒体脑肌病。1991年Leigh综合征的首个致病基因被鉴定[1]。近年来随着基因检测技术的进步,越来越多的基因被报道与Leigh综合征相关。虽然Leigh综合征新致病基因研究进展较快,但关于Leigh综合征的诊断和治疗尚缺乏共识与规范,有必要结合国际最新的研究资料和我国的实际情况,制订符合我国国情的Leigh综合征诊断与治疗的专家共识。2021年12月由中华医学会儿科学分会神经学组、福棠儿童医学发展研究中心神经内科专业委员会和中华儿科杂志编辑委员会共同发起,历时1年8个月制订“Leigh综合征诊断与治疗中国专家共识(2023)”(以下简称本共识),旨在为我国儿童神经专业的临床医生提供更有效指导,规范Leigh综合征的诊断、治疗及管理。
1. 组建共识专家组:本共识由中华医学会儿科学分会神经学组、福棠儿童医学发展研究中心神经内科专业委员会和中华儿科杂志编辑委员会共同发起,首都医科大学附属北京儿童医院临床流行病学与循证医学中心提供技术支持与方法学指导。共识专家组主要邀请有线粒体病临床和基础研究经验的专家参与,同时邀请影像学、基因检测、代谢筛查等相关领域的专家,包括中华医学会儿科学分会神经学组专家41名、组外儿科神经病学专家8名、儿科影像学专家1名、儿科代谢病专家2名、成人神经病学专家1名、线粒体病基础研究专家2名及遗传学专家2名,共57名。
2. 文献综述:组建共识工作组,工作组成员根据国内外Leigh综合征的研究现状,结合临床工作经验,选定临床问题并进行文献检索。计算机检索中国知网、万方全文数据库、维普数据库、中国生物医学文献服务系统、Medline(Via Pubmed)、Embase、The Cochrane Library、Web of science,检索式由“Leigh病”“Leigh综合征”“Leigh disease”“Leigh syndrome”等中、英文相关词汇以逻辑符号组合而成,检索时间为建库至2022年10月,语种限定为中文或英文。根据临床问题,首先通过阅读文献题目及摘要排除不相关的文献。对符合纳入标准的文献,通读全文提取文献证据并综述。前期文献检索发现病例报告、病例系列研究和有限数量的队列研究是主要的证据基础,循证医学证据等级低。因此确定采用德尔菲法制订本共识[2]。共识工作组根据文献检索结果,拟定第1轮德尔菲调查问卷,包括22条推荐意见,涉及Leigh综合征定义、诊断和治疗等各方面。
3. 德尔菲调查:分别于2023年6月和2023年8月进行2轮德尔菲调查。首轮德尔菲调查通过邮件进行,将德尔菲问卷和前期文献检索的证据综述通过邮件发送给专家组各位成员。专家组成员结合文献证据及自身经验,填写德尔菲问卷。每个问题结果分5级,1为强烈反对,2为反对,3为中立,4为同意,5为强烈同意。每个问题后专家均可填写补充意见。若专家填写强烈同意及同意的比例>75%表示该条目达成共识推荐。共识工作组总结首轮德尔菲调查结果,共收回问卷56份。22条推荐意见中共21条达成共识,1条未通过。共识工作组根据专家反馈意见对未达成共识的条目进行修改,对首轮通过的条目进行优化,并新增推荐意见2条。第2轮德尔菲调查采用腾讯视频会议的方式开展。专家组对共识中存在的问题充分讨论,对新增及未达成共识的推荐再次打分。共有42位专家组成员参与第2轮德尔菲调查。最终共24条推荐意见达成共识。
推荐意见1:Leigh综合征是一组由遗传因素导致的原发性线粒体功能障碍,以脑干和(或)基底节对称性受累的头颅影像学改变和相应的临床表现为核心,可合并多系统受累的神经退行性疾病。第1轮德尔菲调查强烈同意+同意98%(55/56)。
Leigh综合征于1951年由英国精神病和神经病理学家Denis Leigh首次报道[3]。1996年,Rahman等[4]首次提出Leigh综合征的临床诊断标准。随着酶学和基因检测技术的发展,2016年Lake等[5]更新了Leigh综合征的诊断标准。2022年北美线粒体疾病联盟共识推荐将Leigh综合征定义为存在发育迟缓或倒退,影像学存在双侧脑干和(或)基底节病灶的原发性线粒体病[6]。
推荐意见2:不再推荐Leigh-like综合征诊断。第1轮德尔菲调查强烈同意+同意84%(47/56)。
根据既往文献,Leigh-like定义为不能完全符合Leigh综合征的诊断条件,多为存在不典型影像、病理或乳酸正常[4, 5]。临床使用过程中,不同医生对此概念理解不同,有的医生把有基底节和(或)脑干对称病灶的非线粒体疾病纳入Leigh-like中,如有机酸代谢障碍;有的医生则认为Leigh-like仍然是线粒体病,但是其影像学可能仅有单侧或不对称脑干和(或)基底节病变;还有医生认为Leigh-like是临床高度怀疑Leigh综合征,但尚未发现致病基因或酶学、病理检查阴性的患者。由此可见,Leigh-like异质性大,内容宽泛,对临床诊断和治疗疾病帮助小,故不再推荐此诊断。
推荐意见3:推荐使用Leigh综合征谱系,定义为一组有与Leigh综合征相同的临床和影像学表现,尚未明确是否为原发性线粒体病的临床影像综合征,诊断需除外获得性疾病(如免疫性脑炎、感染、中毒等)。第2轮德尔菲调查强烈同意+同意95%(40/42)。
对于非典型的Leigh综合征,2020年Rahman和Thorburn[7]首次提出Leigh综合征谱系的概念。2023年同一团队制订了Leigh综合征谱系的诊断标准[8]。根据这一标准,患者若具有Leigh综合征特征性的神经病理表现可直接诊断;否则需要具有与Leigh综合征相同的临床和影像学表现,同时具备线粒体功能障碍的证据(无论原发还是继发性线粒体功能障碍),此标准中不包含基因检查结果。
推荐意见4:Leigh综合征临床症状多样,可累及多系统,多于婴幼儿期起病。当临床表现为发育迟缓、发育倒退和(或)出现基底节、脑干受累的临床症状时,应高度怀疑本病。第1轮德尔菲调查强烈同意+同意100%(56/56)。
前期文献检索发现1篇Meta分析总结至2017年所有文献报道Leigh综合征病例系列的临床特点[9]。此篇Meta分析后又有7篇关于Leigh综合征较大的病例或队列研究发表[10, 11, 12, 13, 14, 15, 16],其中1篇为中国对209例Leigh综合征患儿的队列研究[10]。这8篇研究共报道1 000余例Leigh综合征患者。总体来看,Leigh综合征中位起病年龄在0.25~2.00岁,文献报道2岁前起病占比66%~90%,男女比例相近。神经系统症状方面,发育迟缓是最常见的症状,在以上的研究中占比均≥50%。另外发育倒退,基底节受累引起的肌张力低下、肌张力障碍,脑干受累引起的眼外肌麻痹、吞咽困难、呼吸异常等均为Leigh综合征常见症状,但在不同的研究中占比存在差异。16%~49%的患者可合并癫痫[10, 11, 12, 13, 14, 15, 16],发作类型多样,可为痉挛发作、肌阵挛发作、局灶性发作等,癫痫综合征方面以婴儿癫痫性痉挛综合征最为常见。另外,以上研究均提示除神经系统外,Leigh综合征还可累及心脏、肾脏、肝脏、内分泌系统等多个器官系统。
推荐意见5:头颅影像学检查发现双侧对称性脑干和(或)基底节病变,CT呈低密度,磁共振成像(magnetic resonance imaging,MRI)呈长T2信号,是Leigh综合征诊断的必备条件。第1轮德尔菲调查强烈同意+同意96%(54/56)。
根据多个经典文献中关于Leigh综合征的定义[4, 5, 6],头颅CT或MRI检查发现对称性脑干和(或)基底节病变是Leigh综合征诊断的必备条件。除此之外,部分Leigh综合征患者的病灶可累及大脑皮层、白质、丘脑、小脑、脊髓等部位[10, 11, 12, 13, 14, 15, 16],与其潜在的致病基因不同相关。Leigh综合征病灶在CT上呈低密度,MRI上随病变时期不同,呈现不同的信号特点。急性期病灶肿胀,呈长T1长T2信号,弥散受限,高灌注;亚急性期,部分病灶修复,部分逐渐坏死,呈现混杂信号;慢性期,病灶坏死、局部胶质增生,表现为病灶区域萎缩、囊变、低灌注。
推荐意见6:磁共振波谱分析(magnetic resonance spectroscopy,MRS)发现乳酸峰增高可支持Leigh综合征的诊断,但缺乏敏感性和特异性;推荐对有条件的疑诊Leigh综合征的患儿行此项检查。第1轮德尔菲调查强烈同意+同意98%(55/56)。
Leigh综合征诊断需要有线粒体功能障碍的证据,常规MRI序列不能提供。MRS中乳酸峰增高可作为Leigh综合征诊断的支持证据。虽有文献报道MRS中乳酸峰升高比血乳酸增高诊断线粒体病更敏感[17],但缺乏高质量证据。MRS乳酸峰增高也缺乏疾病特异性,在中枢神经系统肿瘤、缺血缺氧性脑病等其他疾病中均可见到。
推荐意见7:血和(或)脑脊液乳酸增高可支持Leigh综合征诊断,但缺乏敏感性和特异性;此项检测操作简便且费用低,推荐对所有疑诊Leigh综合征的患儿行此项检查。第1轮德尔菲调查强烈同意+同意96%(54/56)。
乳酸检测在Leigh综合征的诊断中缺乏敏感性和特异性。在多项Leigh综合征队列或病例系列研究中[9, 10, 11, 12, 13, 14, 15, 16],Leigh综合征血乳酸增高的比例在68%~74%,脑脊液乳酸增高的比例在64%~80%,故乳酸正常不能除外Leigh综合征。非线粒体疾病如有机酸代谢障碍、药物中毒等亦可引起乳酸增高。血乳酸结果亦受标本采集和处理的限制,如患儿采血过程中使用止血带或剧烈哭闹均可引起乳酸假性升高。因此,血或脑脊液乳酸增高不能确诊Leigh综合征,仅能作为支持证据。而尿液乳酸水平与线粒体疾病的相关性较低[18]。
推荐意见8:在血乳酸升高的情况下,推荐对有条件的疑诊Leigh综合征的患儿测定血乳酸/丙酮酸以鉴别丙酮酸代谢缺陷或氧化磷酸化代谢缺陷导致的Leigh综合征。第1轮德尔菲调查强烈同意+同意96%(54/56)。
在乳酸升高的前提下,测定血乳酸/丙酮酸可帮助鉴别丙酮酸代谢缺陷和氧化磷酸化代谢缺陷,前者通常正常(正常值10∶1~20∶1)或降低,后者则升高[19]。
推荐意见9:推荐对所有疑诊Leigh综合征的患儿行血氨基酸、血酰基肉碱及尿有机酸代谢筛查,以鉴别有特征性血或尿代谢产物的遗传代谢病。第1轮德尔菲调查强烈同意+同意91%(51/56)。
除线粒体病外的其他代谢性疾病,可有与Leigh综合征相似的临床和影像学表现,如甲基丙二酸尿症、尿素循环障碍等。早期进行血和(或)尿代谢筛查可帮助鉴别上述疾病。虽然大部分Leigh综合征患儿的血和(或)尿代谢筛查无特异性,可能仅存在三羧酸循环的中间产物,如苹果酸、延胡索酸等非特异性异常;但仍有少部分基因相关的Leigh综合征血和(或)尿代谢筛查有特征性表现,如CLPB和SERAC1基因变异相关的Leigh综合征尿代谢筛查有3-甲基戊烯二酸升高[20, 21],SUCLA2和SUCLG1基因变异有甲基丙二酸升高[22],ECHS1和HIBCH基因变异有2-甲基-2,3-二羟基丁酸升高[23, 24],ETHE1基因变异则有乙基丙二酸尿等[25]。因此对疑诊Leigh综合征患儿行血和(或)尿代谢筛查,有助于排除血和(或)尿代谢筛查范围内的疾病,并对少部分基因导致的Leigh综合征的诊断有提示作用。
推荐意见10:对于疑诊Leigh综合征患儿,基因检测发现与原发性线粒体能量障碍相关基因的致病性变异可确诊Leigh综合征。推荐对所有疑诊Leigh综合征的患儿行基因检测。第1轮德尔菲调查强烈同意+同意95%(53/56)。
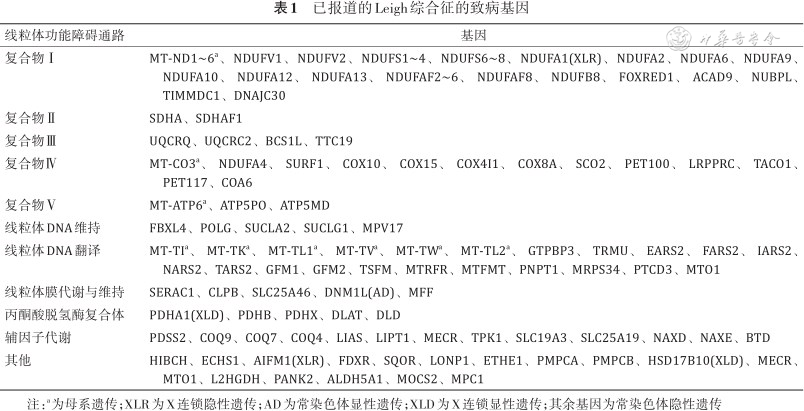
对于疑诊Leigh综合征的患儿,基因检测发现与线粒体原发能量代谢相关基因的致病变异是Leigh综合征的确诊依据。根据Leigh综合征队列和病例系列研究,线粒体DNA(mitochondrial DNA,mtDNA)和核DNA(nuclear DNA,nDNA)变异均可导致Leigh综合征,其中nDNA变异占48%~73%[9, 10, 11, 12, 13, 14, 15, 16]。已有超过100个基因被报道为Leigh综合征的致病基因(表1),常见的致病基因包括MT-ATP6、MT-ND3、MT-ND5、MT-ND6、SURF1、PDHA1、SLC19A3、SUCLA2、ECHS1等[9, 10, 11, 12, 13, 14, 15, 16]。
推荐意见11:推荐首选全外显子组测序(whole exome sequencing,WES)检测nDNA变异;也可选择线粒体病相关nDNA的Panel检测。第2轮德尔菲调查强烈同意+同意98%(41/42)。
在nDNA检测方面,仅少数导致Leigh综合征的nDNA变异有特征性临床表现可提示诊断,从而选择靶向测序,如影像学存在不对称脑室扩大、胼胝体发育不良,生化检查血乳酸/丙酮酸正常或降低高度提示PDHA1基因变异[26]。而在大部分情况下,难以根据临床特点明确需要检测的nDNA。因此,在Leigh综合征中nDNA变异常用线粒体病相关nDNA的基因Panel或WES检测,测序结果需根据美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)指南进行解读[27]。由于不断有新的致病基因被报道,非靶向的WES是更优的选择。对于WES检测阴性的患儿可考虑行全基因组测序(whole genome sequencing,WGS),但其费用高且缺乏统一的数据分析标准,可在有条件的单位探索研究。
推荐意见12:推荐使用基于二代测序的全线粒体基因组测序检测mtDNA变异。推荐首选全血作为检测标本,若未发现致病变异,可进一步检测其他标本(尿、口腔黏膜、肌肉等)的mtDNA(包括点变异和片段缺失),以避免漏检组织特异性变异的可能。第1轮德尔菲调查强烈同意+同意84%(47/56)。
mtDNA检测方面,文献报道Leigh综合征不存在热点变异,因此基于二代测序的全线粒体基因组测序已成为主流。对比一代测序,可显著提高分析点变异、低水平异质性、片段缺失等的可靠性和敏感性。随着年龄的增长,血液中的某些mtDNA变异负荷逐渐降低,因此英国线粒体疾病基因诊断最佳实践指南推荐首选尿液作为成人mtDNA检测的标本,而在儿科血液仍是mtDNA检测的首选标本[28]。需要注意的是,mtDNA变异的分布存在异质性,对于高度怀疑mtDNA变异但全血基因检测阴性者,可进一步检测其他组织(如尿、口腔黏膜、肌肉等)的mtDNA,以避免漏检组织特异性的变异或低水平异质性可能。mtDNA变异的解读可参考美国贝勒医学院临床医学遗传诊断室分类标准进行[29, 30]。
推荐意见13:若有线粒体母系遗传家族史的患儿首先推荐mtDNA检测,否则应同时或先后行mtDNA和nDNA检测。第1轮德尔菲调查强烈同意+同意91%(51/56)。
由于mtDNA检测更为快速和便宜,对于有明确线粒体母系遗传家族史以及经济有困难的患儿可先行mtDNA测序,未发现变异再考虑nDNA测序,否则由于Leigh综合征遗传异质性强,常需同时检测mtDNA和nDNA变异。
推荐意见14:对于基因检测存在意义未明的变异或阴性的疑诊Leigh综合征的患儿推荐肌肉活检行酶活性检测和病理检查,以进一步明确诊断。第1轮德尔菲调查强烈同意+同意93%(52/56)。
在基因检测广泛开展前,组织活检酶学和病理检查是Leigh综合征确诊的主要手段。由于其为有创性检查,随着基因检测的推广,目前组织活检酶学和病理检查主要用于基因检测存在意义未明的变异或阴性的患儿。因骨骼肌是线粒体病常见的受累部位且活检可及性高,故文献中通常选择骨骼肌进行活检。
推荐意见15:当肌肉标本不能获得时,也可皮肤活检行皮肤成纤维细胞酶活性检测。但需注意其阳性率低于肌肉酶学检测。第1轮德尔菲调查强烈同意+同意88%(49/56)。
皮肤活检创伤小,对于不愿行肌肉活检的患儿,可考虑皮肤活检行皮肤成纤维细胞酶活性检测替代[31, 32],但目前大部分研究仍然认为其检测阳性率低于肌肉组织。
推荐意见16:酶活性检测结果受干扰因素多,临床医生需根据有酶活性检测资质的实验室提供的方案行活检前准备、标本采集、保存以及检测结果判读,以降低检测结果的假阴性和假阳性率。第2轮德尔菲调查强烈同意+同意98%(41/42)。
组织酶学检测结果受诸多因素干扰[31],如使用“鸡尾酒”疗法治疗、活检过程中利多卡因渗入肌肉组织、标本没有迅速保存于液氮中等。因此临床医生需根据有线粒体酶活性检测资质的实验室提供的方案进行操作及结果判读,避免错误的结果干扰临床诊断。
推荐意见17:对于疑诊Leigh综合征的患儿,肌肉或皮肤成纤维细胞酶学检测存在呼吸链酶或丙酮酸脱氢酶活性降低,除外继发性因素,可确诊Leigh综合征。第1轮德尔菲调查强烈同意+同意88%(49/56)。
Leigh综合征的酶学检测主要包括呼吸链酶测定和丙酮酸脱氢酶测定,前者最常用的方法为分光光度计法[31],后者为同位素标记法(14C丙酮酸)[33]。但需注意酶活性降低可由继发性病因,如中毒、缺血缺氧等引起。因此,酶活性检测结果必须结合临床进行判断。
推荐意见18:对于疑诊Leigh综合征的患儿,在肌肉标本中发现线粒体病特征性病理表现,除外继发性因素,可确诊Leigh综合征。第1轮德尔菲调查强烈同意+同意96%(54/56)。
线粒体病特征性肌肉病理改变包括改良Gomori三色染色可见破碎红纤维,琥珀酸脱氢酶染色可见破碎蓝染肌纤维,细胞色素C氧化酶染色显示酶活性缺乏。电镜下可见细胞内异常线粒体增多或结构异常,线粒体内可见类结晶包涵体。Leigh综合征肌肉组织病理检查阳性率低,在1项多中心队列研究中[13],仅30.8%患者存在线粒体病特征性的病理改变。与酶学测定一样,所见病理改变应除外继发性病因,结合临床进行判断。
推荐意见19:推荐表2作为Leigh综合征的诊断标准。第1轮德尔菲调查强烈同意+同意96%(54/56)。
推荐意见20:推荐根据Leigh综合征患儿的临床表现和基因检测结果制订个体化、全面的多学科管理和治疗方案。第1轮德尔菲调查强烈同意+同意100%(56/56)。
Leigh综合征目前缺乏有效的治疗方法。除个别基因变异有针对代谢通路缺陷的特异性治疗外,大部分Leigh综合征的治疗以基于多学科的管理和对症支持为主。对患儿合并的癫痫、肌张力障碍、肥厚型心肌病、心脏传导障碍、感音神经性耳聋、眼睑下垂等问题,应及时予以恰当的药物和(或)手术干预。关于癫痫的治疗,目前没有证据表明哪种抗癫痫发作药物单药或联合应用对线粒体病相关的癫痫发作效果更优[34]。在2020年关于原发性线粒体病患者用药安全性的国际专家共识中,推荐丙戊酸仅应在特殊情况下使用且对于POLG基因变异的患者为绝对禁忌,也不应用于合并肝功能损伤的患者[35]。在氨己烯酸使用过程中,部分患儿中出现类似Leigh综合征的影像学改变[36],但大部分患儿无发育倒退、脑干和(或)基底节受累的相关症状,停药后复查影像学病灶消失,在临床工作中应注意鉴别。
推荐意见21:推荐Leigh综合征患儿至康复科制订循序渐进的康复方案,至营养科行营养评估及干预。第1轮德尔菲调查强烈同意+同意98%(55/56)。
运动有助于改善线粒体功能并降低异常线粒体DNA负荷[37, 38]。线粒体病患儿由于最大摄氧量较低易出现运动不耐受,故建议Leigh综合征患儿在专业康复治疗师的指导下制订个性化的康复训练方案,从较低强度和短暂持续时间开始,循序渐进。因Leigh综合征常合并心脏疾病,应注意在开始康复前行心脏方面检查。Leigh综合征易合并营养不良,严重的营养不良也会加重线粒体功能障碍。因此,Leigh综合征患儿应定期到营养门诊进行随访,评估营养状态,并根据患儿的年龄、活动量制订合适的能量摄入方案。对于有吞咽障碍或胃肠蠕动障碍导致经口摄入不足的患儿,建议管饲喂养。
推荐意见22:推荐对临床疑诊Leigh综合征的患儿加用硫胺素10~40 mg/(kg·d)、核黄素10~20 mg/(kg·d)、烟酸10 mg/(kg·d)、生物素5~20 mg/(kg·d)和辅酶Q10 10~30 mg/(kg·d)治疗,以覆盖目前已知的可治疗的Leigh综合征。待基因明确后可根据结果调整治疗方案。第1轮德尔菲调查强烈同意+同意96%(54/56)。
许多呼吸链辅因子及抗氧化剂,包括生物素、硫胺素、核黄素、叶酸、辅酶Q10、维生素E、硫辛酸等,均有报道用于Leigh综合征治疗。患者经常使用上述化合物的组合治疗,因此称“鸡尾酒”疗法。前期文献检索仅发现1篇使用EPI-743治疗Leigh综合征的单臂开放标签的Ⅱa期临床试验[39],此研究纳入10例确诊Leigh综合征患儿,使用EPI-743治疗6个月,所有患儿均出现疾病进展的逆转。其余“鸡尾酒”治疗Leigh综合征均为病例报告,因此“鸡尾酒”治疗Leigh综合征尚缺乏高质量临床证据。由于大部分Leigh综合征患者起病早、病情重且缺乏有效治疗方法,而“鸡尾酒”疗法虽缺乏高质量临床证据,但个别病例有获益且不良反应小,因此目前在线粒体病治疗的国内外指南及专家共识中均推荐使用“鸡尾酒”疗法[31,40],但未对“鸡尾酒”的成分和用量进行推荐。
Leigh综合征中个别基因变异有针对代谢通路缺陷的特异性治疗。TPK1、SLC19A3和SLC25A19基因变异可导致硫胺素代谢障碍,使用大剂量硫胺素治疗有效。其中SLC19A3基因变异是否需合并生物素治疗尚存争议[41, 42]。ACAD9和AIFM1基因变异使用核黄素治疗有效[43, 44]。PDSS2、COQ4、COQ7和COQ9基因变异可导致原发性辅酶Q10缺乏症,使用辅酶Q10治疗有效[45]。PDHA1基因变异导致丙酮酸脱氢酶复合物缺乏症,部分病例硫胺素治疗有效[46]。DLD基因变异的部分患者使用核黄素治疗有效[47]。NAXD和NAXE基因变异应用烟酸治疗有效[48, 49]。BTD基因变异可导致生物素酶缺乏症,使用生物素治疗有效[50]。基于以上文献检索内容,本共识推荐的治疗已覆盖表1中所有已报道基因导致的可治疗的Leigh综合征,推荐剂量参考综述及各基因变异相关的病例报道[41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51]。除烟酸在治疗剂量使用过程中可能出现皮肤潮红、瘙痒、荨麻疹、血清转氨酶升高等不良反应外,其他很少有不良反应出现。待基因明确后可根据结果调整治疗方案。
推荐意见23:推荐对丙酮酸脱氢酶复合物缺乏导致Leigh综合征的患儿使用生酮饮食治疗。第1轮德尔菲调查强烈同意+同意96%(54/56)。
生酮饮食疗法是一种高脂肪、低碳水化合物、合理蛋白质和其他营养素的配方饮食。编码丙酮酸脱氢酶复合物的PDHA1、PDHB、PDHX、DLAT和DLD基因变异导致的Leigh综合征是生酮饮食的绝对适应证[52]。
推荐意见24:推荐对合并难治性癫痫的Leigh综合征患儿,尤其是线粒体复合物Ⅰ缺乏的患儿,排除生酮饮食禁忌后,可考虑生酮饮食治疗。第1轮德尔菲调查强烈同意+同意93%(52/56)。
前期文献检索中,生酮饮食治疗Leigh综合征均为病例报告。2021年的系统综述分析生酮饮食治疗线粒体病的有效性和安全性[53],共有20例基因确诊的使用生酮饮食的线粒体病患儿被纳入(不包括丙酮酸脱氢酶复合物缺乏),8例癫痫患者中7例使用生酮饮食后癫痫发作得到控制,5例肌肉存在mtDNA单一或多重缺失的患者使用生酮饮食后出现横纹肌溶解,据此,该作者认为生酮饮食治疗线粒体病数据太少,无法提供一般性建议。对于线粒体病合并难治性癫痫的患者,排除生酮饮食的禁忌证后,可考虑生酮饮食。根据2018年儿童生酮饮食治疗癫痫的最佳临床管理[54],对于丙酮酸脱氢酶复合物以及线粒体复合物Ⅰ缺乏的癫痫患儿,生酮饮食有更多获益(>70%),推荐应尽早使用。
本共识基于德尔菲调查问卷对Leigh综合征的定义、诊断和治疗给出了24条推荐意见。基因的检测技术日新月异,几乎每年都有新的Leigh综合征的致病基因被报道,因此本共识需结合最新研究进展进行定期更新。从目前证据检索结果可以看出,Leigh综合征总体证据质量低,今后需要多中心合作共同推进Leigh综合征的临床研究,从而产生高质量的文献证据。另外,目前Leigh综合征缺乏有效治疗方法,总体预后差,多项Leigh综合征队列研究或病例系列研究显示至末次随访时病死率在17%~39%,中位死亡年龄2.4~4.0岁[9, 10, 11, 12, 13, 14]。已有一些基因治疗正在动物研究阶段,有几项药物已进入临床试验阶段,有望获得美国食品药物管理局批准。期待这些药物能尽快应用于临床,改善Leigh综合征患儿预后。
参与本共识制订的专家组成员(按单位及姓名拼音排序):安徽医科大学第一附属医院(吴德);北京大学第一医院(包新华、季涛云、姜玉武、马祎楠、戚豫、王朝霞、吴晔、杨艳玲);北京大学人民医院(秦炯、杨志仙);沧州市人民医院(王荣);重庆医科大学附属儿童医院(洪思琦);复旦大学附属儿科医院(王艺、周水珍);福建医科大学附属协和医院(胡君);广西医科大学第一附属医院(韩蕴丽);哈尔滨医科大学附属第六医院(王春雨);河北省儿童医院(孙素珍);河南省儿童医院(王媛);河南省人民医院(高丽);华中科技大学同济医学院附属同济医院(刘艳);华中科技大学同济医学院附属武汉儿童医院(刘智胜、孙丹);吉林大学第一医院(梁建民);江西省儿童医院(吴华平);解放军总医院第一医学中心(杨光、邹丽萍);兰州大学第二医院(陈永前);南京市儿童医院(郑帼);内蒙古医科大学附属医院(杨光路);宁夏回族自治区人民医院(卞广波);日本松本生命科学研究所(张春花);山东大学齐鲁医院(李保敏);山西省儿童医院(韩虹);上海交通大学医学院附属上海儿童医学中心(王纪文);上海交通大学医学院附属新华医院(李玲);深圳市儿童医院(廖建湘);首都儿科研究所附属儿童医院(陈倩、杨健);首都医科大学附属北京儿童医院(程华、方方、任晓暾);四川大学华西第二医院(甘靖、罗蓉);天津市儿童医院(张玉琴);温州医科大学(方合志);西藏自治区人民医院(赵蓉);新疆维吾尔自治区人民医院(孙岩);新乡医学院第三附属医院(韩金芬、王家勤);云南省第一人民医院(汤春辉);浙江大学医学院附属儿童医院(高峰);郑州大学第一附属医院(糕志红);中国医科大学附属盛京医院(张俊梅);中南大学湘雅医院(彭镜);遵义医科大学附属医院(田茂强)
共识制订工作组成员(按姓名拼音排序):首都医科大学附属北京儿童医院(徐超龙、周季、邹颖)
共识制订方法学指导专家(按姓名拼音排序):首都医科大学附属北京儿童医院(刘雅莉、聂晓璐)